R语言学习笔记17
Data Summarisation
数据整理
拆分数据列
看下面的数据。分隔方式不同
d.sep <- read_csv( |
| testid | succ/total |
|---|---|
| 1 | 1/10 |
| 2 | 3/5 |
| 3 | 2/8 |
用tidyr::separate()可以将这样的列拆分为各自的变量列,如
d.sep %>% |
| testid | succ | total |
|---|---|---|
| 1 | 1 | 10 |
| 2 | 3 | 5 |
| 3 | 2 | 8 |
其中into指定拆分后新变量名, sep指定分隔符, convert=TRUE要求自动将分割后的值转换为适当的类型。 sep还可以指定取子串的字符位置, 按位置拆分各个子串。
选项extra指出拆分时有多余内容的处理方法, 选项fill指出有不足内容的处理方法。
dbpa <- read_csv( |
# 用separate将var的值分为两列 |
| sex | var | avg |
|---|---|---|
| m | s | 118 |
| m | d | 85 |
| f | s | 115 |
| f | d | 83 |
然后再将男女分为两列
dbpa2 <- dbpa1 %>% |
| var | m | f |
|---|---|---|
| s | 118 | 115 |
| d | 85 | 83 |
将男女分为两行
dbpa3 <- dbpa1 %>% |
| sex | s | d |
|---|---|---|
| m | 118 | 85 |
| f | 115 | 83 |
函数extract()可以按照某种正则表达式表示的模式从指定列拆分出对应于正则表达式中捕获组的一列或多列内容。 例如,下面的数据中factors水平AA, AB, BA, BB实际是两个因子的组合, 将其拆分出来:
dexp <- tibble( |
| design | response |
|---|---|
| AA | 120 |
| AB | 110 |
| BA | 105 |
| BB | 95 |
dexp1 <- extract(dexp, design, into = c('f1','f2'), regex="(.)(.)") |
| f1 | f2 | response |
|---|---|---|
| A | A | 120 |
| A | B | 110 |
| B | A | 105 |
| B | B | 95 |
合并数据列
先将d.sep分隔
d.sep1 <- d.sep %>% |
然后使用unite合并同一行的两列或多列内容
d.sep2 <- unite(d.sep1, ratio, succ, total, sep = ":") |
| testid | ratio |
|---|---|
| 1 | 1:10 |
| 2 | 3:5 |
| 3 | 2:8 |
unite()的第一个参数是要修改的数据框, 这里用管道%>%传递进来, 第二个参数是合并后的变量名(ratio变量), 其它参数是要合并的变量名,sep指定分隔符。 实际上用mutate()、paste()或者sprintf()也能完成合并。
# 用paste合并 |
| testid | ratio |
|---|---|
| 1 | 1:10 |
| 2 | 3:5 |
| 3 | 2:8 |
数据框纵向合并
矩阵或数据框要纵向合并,使用rbind函数即可。 dplyr包的bind_rows()函数也可以对两个或多个数据框纵向合并。 要求变量集合是相同的,变量次序可以不同。
比如,有如下两个分开男生、女生的数据框:
d3.class <- filter(select(d.class,name, sex, age),sex=="M") |
合并如下:
knitr::kable(bind_rows(d3.class,d4.class)) |
| name | sex | age |
|---|---|---|
| Alfred | M | 14 |
| Duke | M | 14 |
| Guido | M | 15 |
| James | M | 12 |
| Jeffrey | M | 13 |
| John | M | 12 |
| Philip | M | 16 |
| Robert | M | 12 |
| Thomas | M | 11 |
| William | M | 15 |
| Alice | F | 13 |
| Becka | F | 13 |
| Gail | F | 14 |
| Karen | F | 12 |
| Kathy | F | 12 |
| Mary | F | 15 |
| Sandy | F | 11 |
| Sharon | F | 15 |
| Tammy | F | 14 |
# 与rbind类似 |
| name | sex | age |
|---|---|---|
| Alfred | M | 14 |
| Duke | M | 14 |
| Guido | M | 15 |
| James | M | 12 |
| Jeffrey | M | 13 |
| John | M | 12 |
| Philip | M | 16 |
| Robert | M | 12 |
| Thomas | M | 11 |
| William | M | 15 |
| Alice | F | 13 |
| Becka | F | 13 |
| Gail | F | 14 |
| Karen | F | 12 |
| Kathy | F | 12 |
| Mary | F | 15 |
| Sandy | F | 11 |
| Sharon | F | 15 |
| Tammy | F | 14 |
横向合并
为了将两个行数相同的数据框按行号对齐合并, 可以用基本R的cbind()函数或者dplyr包的bind_cols()函数。
实际数据往往没有存放在单一的表中, 需要从多个表查找数据。 多个表之间的连接, 一般靠关键列(key)对准来连接。 连接可以是一对一的, 一对多的。 多对多连接应用较少, 因为多对多连接是所有两两组合。
在规范的数据库中,每个表都应该有主键, 这可以是一列,也可以是多列的组合。 为了确定某列是主键, 可以用count()和filter(),如:
# 可以用以下语句查看tibble数据某列是否有重合的数据 |
没有发现重复出现的name, 说明d.class中name可以作为主键。
d1.class <- d.class %>% |
# 查看语句的功能 |
此处的!(name %in% 'Becka'))表示的就是在name中去掉Becka。
用dplyr包的inner_join()函数将两个数据框按键值横向合并, 仅保留能匹配的观测。因为d1.class中丢失了Becka的观测, 所以合并后的数据框中也没有Becka的观测:
inner_join(d1.class, d2.class) %>% |
Joining, by = “name”
| name | sex | age | height | weight |
|---|---|---|---|---|
| Alice | F | 13 | 56.5 | 84 |
| Gail | F | 14 | 64.3 | 90 |
| Karen | F | 12 | 56.3 | 77 |
原来的前三行包含Becha的数据。
横向连接自动找到了共同的变量name作为连接的键值, 可以在inner_join()中用by=指定键值变量名, 如果有不同的变量名, 可以用by = c(“a”=“b”)的格式指定左数据框的键值a与右数据框的键值b匹配进行连接。
cbind也类似
# 取出几列值,来操作列合并 |
| name | sex | add | weight | height |
|---|---|---|---|---|
| Alice | F | 1 | 84 | 56.5 |
| Becka | F | 2 | 98 | 65.3 |
| Gail | F | 3 | 90 | 64.3 |
left_join()按照by变量指定的关键列匹配观测, 左数据集所有观测不论匹配与否全部保留, 右数据集仅使用与左数据集能匹配的观测。 不指定by变量时, 使用左、右数据集的共同列作为关键列。 如果左右数据集关键列变量名不同, 可以用by=c(“左名”=“右名”)的格式。
类似地, right_join()保留右数据集的所有观测, 而仅保留左数据集中能匹配的观测。 full_join()保留所有观测。 inner_join()仅保留能匹配的观测。
left_join()将右表中与左表匹配的观测的额外的列添加到左表中。 如果希望按照右表筛选左表的观测, 可以用semi_join(), 函数anti_join()则是要求保留与右表不匹配的观测。
标准化
设x是各列都为数值的列表(包括数据框和tibble)或数值型矩阵, 用scale(x)可以把每一列都标准化, 即每一列都减去该列的平均值,然后除以该列的样本标准差。 用scale(x, center=TRUE, scale=FALSE)仅中心化而不标准化。
|
d.class %>% |
注意在管道操作中某个操作除了被传递的第一自变量外没有其它自变量时, 可以不写函数调用的空括号()。
28 基本R绘图
R的基本绘图功能有两类图形函数: 高级图形函数, 直接针对某一绘图任务作出完整图形,如 如lattice、ggplot2绘图系统; 低级图形函数,在已有图形上添加内容。 具备有限的与图形交互的能力(函数locator 和identify)。
常用的高级图形
条形图
d.cancer数据框包含了肺癌病人放疗的一些数据, 从cancer.csv读入:
d.cancer <- readr::read_csv("cancer.csv", locale = locale(encoding="GBK")) |
统计男女个数并用条形图表示:
res1 <- table(d.cancer[,'sex']);print(res1) |
barplot(res1) |
可以增加标题,采用不同的颜色:
barplot(res1, main = "性别分布", col = c("brown2", "turquoise1")) |

R函数colors()可以返回R中定义的用字符串表示的六百多种颜色名字。 如
head(colors(),8) |
下面的函数可以用来挑选颜色, 鼠标点击画出的颜色就可以挑选, 结果返回挑选出的颜色名:
select.colors <- function(){ |
# 在plots窗口中点选颜色 |

用width选项与xlim选项配合可以调整条形宽度,如
barplot(res1, width = 0.5, xlim=c(-3,5), main = "性别分布", col=c("brown2", "aquamarine1")) |
按性别与病理类型交叉分组后统计频数,结果称为列联表:
res2 <- with(d.cancer, table(sex,type)) |
| 鳞癌 | 腺癌 | |
|---|---|---|
| F | 4 | 9 |
| M | 18 | 3 |
用分段条形图表现交叉分组频数, 交叉频数表每列为一条:
barplot(res2, legend=T) |

用并排条形图表现交叉分组频数, 交叉频数表每列为一组:
barplot(res2, beside=TRUE, legend=TRUE) |
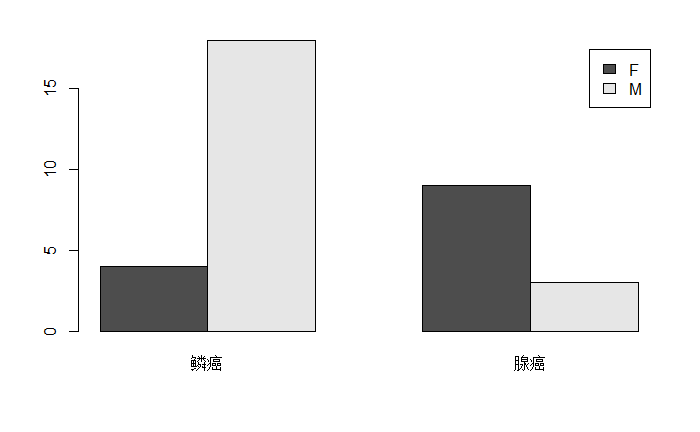
增加标题,指定颜色,调整图例位置,调整条形宽度:
barplot(t(res2), beside = T, legend=T, main="不同性别的病人种类", ylim=c(0,20),xlim=c(-1,6), width=0.6,col=c("brown2","aquamarine1")) |

直方图和密度估计图
# 用hist作直方图以了解连续取值变量分布情况 |
可以用main=、xlab=、ylab=等选项, 可以用col=指定各个条形的颜色,如:
hist(x, col=rainbow(14),main = "normal distribution", xlab="", ylab="频数") |

函数density()估计核密度。 下面的程序作直方图, 并添加核密度曲线:
tmp.dens <- density(x) |

参数说明:
main:标题
xlab,ylab:x,y轴说明
xlim,ylim:x,y轴的刻度界限
col:图的颜色
lines()函数必须和plot()函数配合才能使用,先用plot()函数画出一个图形,再用lines()函数加上其他线条。
盒形图
在R中创建盒形图的基本语法是:
boxplot(x, data, notch, varwidth, names, main) |
以下是使用的参数的描述 -
- x - 是向量或公式。
- data - 是数据帧。
- notch - 是一个逻辑值,设置为
TRUE可以画出一个缺口。 - varwidth - 是一个逻辑值。设置为
true以绘制与样本大小成比例的框的宽度。 - names - 是将在每个箱形图下打印的组标签。
- main - 用于给图表标题。
# 使用R中已经存在的数据集mtcars来创建基本的boxplot |
mpg(每加仑英里)和cyl(气缸数)列之间的关系创建一个盒形图。
getwd() |

盒形图与凹口
可以绘制带有凹槽的盒形图,以了解不同数据组的中位数如何相互匹配。
# plot chart with notch |

盒形图可以简洁地表现变量分布,如
with(d.cancer, boxplot(v0)) |

with(data, expr, …)函数用于在一个从data构建出的环境中运行R表达式。
其中中间粗线是中位数, 盒子上下边缘是和分位数, 两条触须线延伸到取值区域的边缘。
盒形图可以很容易地比较两组或多组,如
with(d.cancer, boxplot(v0 ~ sex)) |

也可以画若干个变量的并排盒形图,如
with(d.cancer, |

 微信
微信 支付宝
支付宝
