每日外闻42
Smoke signals in the DNA of normal lung cells
吸烟者的正常细胞中有更高的突变负担,这类似于肺癌患者的肺细胞突变。出人意料的是,以前吸过烟,现在戒烟了的人,其肺部的细胞有很大一部分是正常的,几乎与正常细胞一致。
According to the World Health Organization, there are 1.1 billion smokers worldwide and an estimated 1.8 million deaths from lung cancer annually. Lung cancer caused by smoking can take decades to arise, and smokers have up to a 30-fold higher risk of developing the disease than do non-smokers1. Carcinogenic components of tobacco smoke promote lung cancer by causing DNA damage that can lead to mutations through known mechanisms, but what the initial consequences of smoking are for healthy lung cells is poorly understood. Writing in Nature, Yoshida et al.2 report the mutational profiles of 632 healthy lung cells obtained from whole-genome sequencing of biopsied tissue from 16 individuals: children, adults, non-smokers, current smokers and ex-smokers. The authors analysed the frequency and properties of the mutations present, how they differed according to age and smoking status, and how these mutations related to those found in a type of lung cancer called squamous-cell carcinoma.
The authors dissociated cells from lung tissue (Fig. 1) and isolated a type of epithelial cell called a basal cell (which can self-renew). Growing single cells into cellular colonies allowed the authors to determine the DNA sequence of the given original cell. A potential caveat of the study is that, although the authors obtained the genome sequences of hundreds of single cells, the number of individuals with each different smoking status was relatively small. The authors report that the number of single nucleotide (point) mutations increased with age — for each extra year of life, about 22 additional such mutations were found per cell.
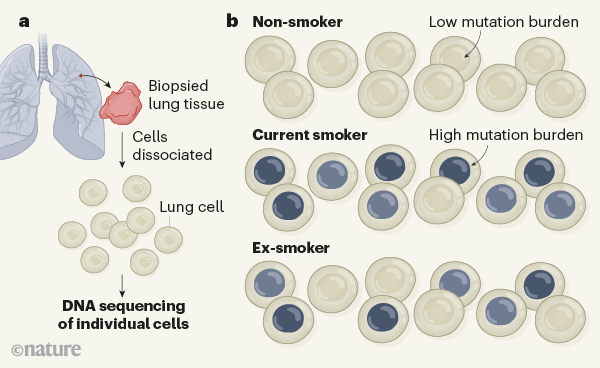
Figure 1 | Mutational burdens in normal human lung cells. Yoshida et al.2 analysed the pattern of mutations in healthy lung tissue in non-smokers, current smokers and ex-smokers. a, Using biopsied lung tissue, the authors determined whole-genome sequences corresponding to single cells. b, The cells of the non-smoking individuals had few mutations. By contrast, current smokers had a high proportion of cells with a large number of mutations (grey; darker colour indicates more mutations), and many of these mutations were of a type predominantly found in smokers. Compared with non-smokers, smokers also had greater variability in the mutational load between the different cells of a given individual. Surprisingly, the authors found that five out of six ex-smokers had a substantial fraction (20–50%) of cells that had low numbers of mutations and had hardly any smoking-associated mutational signatures. How these cells arise is a mystery — Yoshida et al. speculate that they are generated from a population of as-yet-unknown stem cells.
However, being a former smoker added another 2,330, and being a current smoker added 5,300 point mutations per cell on average, confirming the mutational potency of smoking. Smokers’ genomes also had extensive examples of other types of alteration, such as insertion or deletion mutations. The number of mutations in different cells from the same individual could vary by tenfold in smokers, a much higher variability than was found in non-smokers. The stage of the cell cycle at which a cell is exposed to carcinogenic agents might affect how effectively DNA damage is repaired before DNA replication, which could offer an explanation for this high variability.
Yoshida and colleagues examined the mutations in individual cells using previously developed algorithms to focus on all the types of sequence alteration possible (for example, mutation of the DNA base adenine to cytosine, guanine or thymine) and also to assess the bases on either side of a mutated base. Such analysis identifies specific patterns (mutational signatures) that have been used before to characterize the genomes of tumour cells3.
The authors report that the presence of certain mutational signatures increased with age and did not seem to be affected by smoking. These included a signature attributed to natural processes whereby the loss of an amino group in a modified cytosine (termed 5-methylcytosine) changes the base to a thymine. The most common mutational signature in all the samples was one that is rich in cytosine-to-thymine and thymine-to-cytosine mutations. The presence of this signature increased with age and was more common in people with a history of smoking. The underlying processes driving these mutations are unknown. The most common smoking-dependent signature consisted of guanine-to-thymine mutations, a signature that is characteristic of most smoking-associated lung cancers4–7.
Lung cancers have some of the highest mutation frequencies of all tumour types8; however, it is thought that only a small number of tumour-promoting (driver) mutations need to occur in a single cell to kick off malignant growth. Given the high mutational burden and the specific smoking-associated mutational signatures found in smokers’ healthy epithelial cells, Yoshida and colleagues examined whether these mutations affected crucial genes that are relevant for cancer growth.
Indeed, they found cells that had acquired mutations in genes, including TP53 and NOTCH1, that are driver mutations in squamous-cell carcinomas. These driver mutations were more common in the lung cells of smokers than in those of non-smokers. Some cells even had as many as three driver mutations. However, we do not know how many of these mutations (and in what combination) are required for human lung cancer to develop. Specific TP53 mutations were found in multiple cells from the same individual, suggesting that these mutations occur early, that cells with the mutation proliferate, or both — similar to what has been observed for sun-exposed healthy human skin9.
The higher risk of lung cancer in ex-smokers compared with non-smokers is reflected in their high mutation burden and the signature of smoking-associated mutations in most of their lung cells (similar to the cellular profile of current smokers). Although ex-smokers have a high risk of developing lung cancer, their risk is reduced compared with that of current smokers, and this lowering depends on the length of time of smoking cessation1. Why this is the case has been hard to explain. However, perhaps the most surprising result of Yoshida and colleagues’ work might offer a clue: in 5 out of 6 ex-smokers, 20–50% of the cells had a low mutation burden that was similar to the profile of non-smokers of the same age range (Fig. 1).
These near-normal cells in ex-smokers had a low frequency of smoking-dependent mutational signatures. Moreover, compared with the ex-smokers’ highly mutated cells, these near-normal cells had longer versions of DNA structures called telomeres, which are found at the ends of chromosomes. Telomere length shortens with each cell division; thus, long telomeres suggest that these cells had not undergone many divisions. The authors speculate that these cells might have arisen comparatively recently from divisions of proposed previously dormant (quiescent) stem cells. However, whether such cells exist in human lungs is unknown.
DNA damage can generate a mutation during DNA replication. Therefore, if a population of non-dividing stem cells exists in the human lung, even if exposed to carcinogenic agents, perhaps such cells might avoid incurring mutations if DNA damage is eventually repaired in the absence of division. But the lack of knowledge about these proposed long-lived stem cells and information about the longevity of the different cell types in the human lung make it difficult to explain what occurred in these ex-smokers’ cells with few mutations.
Why do ex-smokers still have a substantial fraction of highly mutated cells that can proliferate, at least when grown in vitro? Any short-lived cells that were exposed to carcinogens during their proliferation should have vanished many years after the cessation of smoking. This raises the question of whether there are long-lived differentiated cells in the lung that carry a high mutational burden, and whether these cells can resume proliferation, perhaps because of the plasticity (the ability to change cellular identity) of lung cells10. A future challenge will be to understand the cell biology of the mechanisms underlying these observations. Perhaps one day it will be possible to develop ways to boost the population of lung cells with few mutations in ex-smokers.
Yoshida and colleagues’ study has broadened our understanding of the effects of tobacco smoke on normal epithelial cells in the human lung. It has shed light on how the protective effect of smoking cessation plays out at the molecular level in human lung tissue and raises many interesting questions worthy of future investigation.
Reference:
https://www.nature.com/articles/d41586-020-00165-7
See you tomorrow
 微信
微信 支付宝
支付宝